Die Alzheimer-Erkrankung - ein rätselhafter Kriminalfall (Teil II)

Folge 20: Aktuelle Chemie 2019 - Medizin und Gesundheit
Unser Gehirn besteht aus einem riesigen Netzwerk aus rund hundert Milliarden Nervenzellen, den Neuronen. Über Kontaktstellen, die sogenannten Synapsen, sind die einzelnen Neuronen miteinander verbunden, tauschen Informationen aus und speichern diese ab. In einem von Alzheimer-Demenz befallenen Gehirn sind diese Verbindungen gekappt. Je nachdem, in welchem Areal das passiert, sind zum Beispiel das Kurzzeitgedächtnis oder das Sprachvermögen von den Schäden betroffen. Wie in Teil I bereits erläutert, gibt es bei der Erforschung der Ursache der Erkrankung noch keine eindeutigen Antworten. Was man jedoch feststellen kann, sind drei anomale Veränderungen im Gehirn, die schon der Arzt Alois Alzheimer 1906 beobachtete: Zum einen die sogenannte „Plaques“, die aus polymerisierten Amyloid-Beta-Proteinen (kurz Aβ) bestehen und die sich in den betroffenen Arealen in der Nähe von Neuronen häufen. Dazu kommt ein weiteres Protein, das sich zunächst innerhalb der Neuronen zusammenlagert und weitere Ablagerungen bildet: das sogenannte Tau-Protein. Ebenso charakteristisch sind die Anzeichen einer chronischen Entzündung im Gehirn, die sich durch Veränderungen von Mikroglia-Zellen, Abwehrzellen des Gehirns, zeigen. Aufbauend auf diese drei Beobachtungen gibt es zahlreiche wissenschaftliche Ansätze, die versuchen zu ergründen, wie es zu dem jeweiligen Zustand kommt und wie man ihn verhindern bzw. umkehren kann.
Eine hochkomplexe Aufgabe
In einem ist sich die Wissenschaft bereits einig: Die One-Fits-All-Lösung wird es nicht geben. „Das liegt daran, dass es sich um eine äußerst heterogene Erkrankung handelt“, erklärt Prof. Dr. Michael Heneka, Direktor der Klinik für Neurodegenerative Erkrankungen und Gerontopsychiatrie der Universität Bonn. „Das heißt, dass die Erkrankung in jedem Hirnareal unterschiedlich weit fortgeschritten sein kann. Man kann sich das vorstellen wie einen Staffellauf mit mehreren Teams. Beispielsweise könnte Aβ der erste Läufer sein, der den Staffelstab an die Immunantwort weitergibt und diese wieder an den nächsten ‚Läufer‘, z.B. das Tau-Protein. Nun laufen in einem Stadion jedoch mehrer Teams im gleichen Rennen, wenn auch unterschiedlich schnell. Man stelle sich vor, dass jedes Hirnareal ein Team repräsentiert: Versucht man nun in einem Areal, in dem der Staffelstab bereits an weitergegeben wurde, das Aβ zu bekämpfen, wird man vermutlich keinen Erfolg haben.“ Dies könne ein Grund dafür sein, dass die bisher auf Aβ fokussierten Therapie-Ansätze gescheitert seien – sie wären schlichtweg zu spät gekommen. Das müsse jedoch nicht heißen, dass sie komplett wirkungslos seien, nur eben möglicherweise in einem anderen Stadium der Erkrankung ihren Einsatz hätten. „Wenn wir nun das ganze Gehirn überall gleichzeitig schützen wollen, müssten unterschiedliche Therapiestrategien zur gleichen Zeit eingesetzt werden. Eine wirksame Therapie müsste entsprechend eine hochkomplexe und für den einzelnen Patienten sehr individuelle Zusammensetzung haben. So weit ist man in der Medizin jedoch noch lange nicht. Aber auch die Wissenschaft arbeitet sozusagen in Staffel-Teams – und jeder Erfolg trägt dazu bei, dass wir dem Ziel näher kommen.“
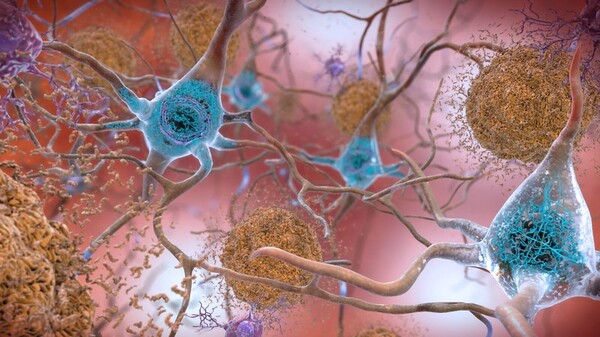
Im Gehirn von Alzheimer-Patienten finden sich Aβ-Plaques (hier braun), die sich zwischen den Neuronen ansammeln und die Zellfunktionen stören. Zudem sammeln sich Tau-Fibrillen innerhalb der Neuronen an (hier blau), die die Synapsen-Kommunikation stören. Bild: National Institute on Aging, NIH
Das Tau-Protein im Visier
Eine führende Forschergruppe am Deutschen Zentrum für Neurodegenerative Erkrankungen (DZNE) zum Thema Tau-Protein und dessen Rolle bei der Alzheimer-Erkrankung befindet sich unter der Leitung von Prof. Dr. Eckhard Mandelkow und seiner Frau Dr. Eva-Maria Mandelkow. Das Tau-Protein ist ein Protein, das an Mikrotubuli, einem wichtigen Bestandteil des Zellskeletts, bindet und an ihrem korrekten Zusammenbau sowie deren Stabilisierung beteiligt ist. Auch bei der Weiterleitung von Informationen und der Nährstoffversorgung spielt es eine Rolle. Wenn das Tau-Protein verstärkt Phosphatgruppen bindet, Hyperphosphorylierung genannt, verändert es seine Wechselwirkungen mit anderen Zellkomponenten und es kann es zu einer unkontrollierten Polymerisation, also Verkettung der Proteine, kommen. Diese Ketten lagern sich wiederum zu Bündeln zusammen, den für Alzheimer-Patienten charakteristischen Tau-Fibrillen (auch Alzheimer-Fibrillen genannt). Das Zellskelett verliert seine stützenden Helfer und wird instabil; auch die Informationsweiterleitung ist gestört. Nach und nach verliert die Zelle ihre Funktion und stirbt. Das Mandelkow-Team untersucht diese Zusammenhänge. „Wir untersuchen Maus-Modelle der Alzheimer-Demenz, bei denen die pathologischen Veränderungen durch eine Mutation im Tau-Protein ‚angeschaltet‘, aber auch wieder abgeschaltet werden können“, berichtet Eckhard Mandelkow. „Bei diesen Mäusen haben wir dann untersucht, wie sich die typischen Störungen in Gedächtnis und Verhalten entwickeln, und ob sie sich durch Ausschalten des dafür verantwortlichen Gens auch wieder abstellen lässt. Die unerwartete Antwort war: Ja, die Maus, die davor deutliche Alzheimer-Symptome zeigte, wird nach dem Abschalten des mutierten Tau-Gens auch wieder vollkommen gesund.“ Diesen Erfolg müsse man differenziert betrachten, da es sich um gentechnisch veränderte Mäuse handele und sich die Ergebnisse nicht einfach auf den Menschen übertragen ließen. Dennoch ein vielversprechender Ausgangspunkt.
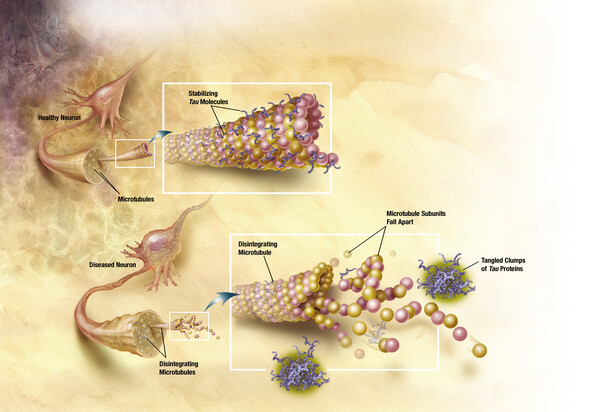
Gesundes Neuron mit vom Tau-Protein gestützten Mikrotubuli (oben) und erkranktes Neuron mit zerfallenden Mikrotubuli und aggregierenden Tau-Proteinen (unten). Bild: ADEAR: "Alzheimer's Disease Education and Referral Center, a service of the National Institute on Aging.", TANGLES HIGH, als gemeinfrei gekennzeichnet, Details auf Wikimedia Commons
„Angetrieben von der Erkenntnis, welch große Rolle Tau im Alzheimer-Prozess spielen kann, haben wir einen weiteren, Gen-unabhängigen Ansatz entwickelt. Und zwar haben wir Alzheimer-Mäusen mit Tau-Fehlfaltung einen Wirkstoff verabreicht, der bestimmte Signal-Proteine auf der Oberfläche von Neuronen blockiert. Auch diese Mäuse erholen sich wieder von der Erkrankung – die Neuronen nehmen ihre Arbeit wieder auf und die kognitiven Fähigkeiten der Maus verbessern sich.“ Der Zusammenhang zwischen extrazellulären Signalen und der intrazellulären Antwort (Funktionen des Tau-Proteins und Re-Aktivierung der Synapsen) ist noch nicht geklärt. Aber das Beispiel zeigt, dass es durchaus möglich ist, kognitive Störungen wieder rückgängig zu machen.
Um die Tau-Pathologie ranken sich aktuell noch viele weitere Ansätze, die z.T. auch schon in klinischen Studien laufen. So wurde beispielsweise beobachtet, dass das fehlgefaltete Tau-Protein über Exosomen, kleine Vesikel, die von einer Zelle zur anderen übertragen werden, von einer erkrankten Zelle an eine gesunde weitergegeben werden. Dieses sorgt dafür, dass auch dort die Fehlfaltung und Aggregation einsetzt. Der therapeutische Ansatz einiger Pharma-Unternehmen liegt darin, das Tau-Protein beim Verlassen der Zelle mit einem Antikörper abzufangen und so die Ausbreitung zu verhindern. Im Mausmodell hat dies bereits erste Erfolge gezeigt; Ergebnisse an menschlichen Patienten stehen noch aus.
Mikroglia: Die gehirneigene Abwehr nutzen
Ein weiteres Forschungsfeld, mit dem sich unter anderem Michael Heneka beschäftigt, ist die chronische Entzündung, die sich im Gehirn von vielen Alzheimer-Patienten feststellen lässt. Ausgelöst wird diese von Mikroglia. „Schädliche Bakterien tragen auf ihrer Zelloberfläche beta-fehlgefaltete Amyloide, sogenannte curly fibres“, erklärt Heneka, der am DZNE die Forschungsgruppe Neuroinflammation leitet. „Mikroglia sind ausgestattet mit Mustererkennungsrezeptoren, die diese curly fibres erkennen. Haben sie einen Eindringling detektiert, produzieren sie Zytokine, also Zellgifte, mit denen sie die Erreger töten. Im Anschluss nehmen sie die toten Zellen auf und verstoffwechseln diese. Beim Alzheimer ist die Situation nun ein wenig anders: Es gibt zwar überall Signale für einen Bakterienbefall durch das Beta-Amyloid, doch liegt in Wirklichkeit kein Befall vor. Dadurch resultiert eine chronische Entzündung, die über die Zeit zunächst die Funktion und schließlich auch die Struktur der Nervenzellen negativ beeinflusst.“ Gleichzeitig setzten die Mikroglia in ihrem Angriffsmodus eine Proteinmischung frei, die Aβ-Proteine einfängt und aggregiert und somit die Plaque-Bildung verstärkt.
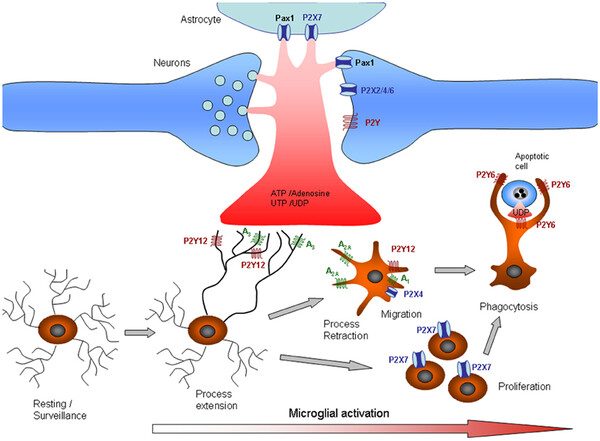
Signalkaskade zur Aktivierung von Mikroglia. Bild: Frontiers in cellular neuroscience, Purinergic signalling Microglia, CC BY 3.0
In einer aktuellen in der Zeitschrift Nature veröffentlichen Studie konnte die Forschergruppe um Michael Heneka am Uniklinikum und DZNE Bonn außerdem zeigen, dass aktivierte Mikroglia auch die Tau-Hyperphosphorylierung beeinflussen. „Wir haben herausgefunden, dass ein bestimmtes Imflammasom namens NLRP3, ein Proteinkomplex des angeborenen Immunsystems, sich in aktivierten Mikroglia ansammelt. Es veranlasst die Mikroglia dazu, bestimmte Stoffe auszustoßen, die wieder andere Proteine aktivieren, die letztendlich zur Fehlfaltung von Tau führen können. Also haben wir untersucht, ob die Verringerung von NLRP3 in erkrankten Maus-Mikroglia die Ausbreitung der Tau-Pathologie verhindert und konnten einen positiven Effekt feststellen. Diesen Ansatz verfolgen wir nun weiter.“
Auf Basis der Erkenntnisse rund um die Rolle der Mikroglia hat sich ebenfalls ein breites Forschungsfeld entwickelt, das inzwischen zahlreiche unterschiedliche Ansätze verfolgt.
Hinweis: Dieser Beitrag erhebt keinen Anspruch auf Vollständigkeit, sondern stellt lediglich eine kleine Auswahl aktueller Ansätze der Alzheimerdemenzforschung dar.

Kommentare
Keine Kommentare gefunden!